The Wittig Reaction – Formal Report
The Wittig Reaction – Formal Report
By: Alexander Davies
Introduction
Alkenes are important initial building blocks in many organic synthesis routes, as shown by Scheme 1.1 It is paramount, therefore, that we have the ability to synthesize alkenes from other readily available, cheap material.
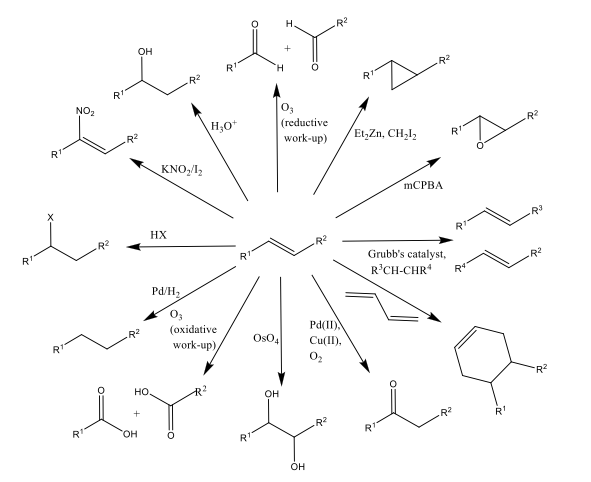
Scheme 1: Examples of one-step reactions from an alkene starting material. This illustrates the huge diversity of olefin products; and as such the need to be stereoselective in how one can synthesise said functionality within a molecule.
Alkene synthesis via dehydration is relatively simple and results in high yields of a mixture of alkene isomers.2 However, this method does not allow one to be completely selective in producing a single alkene structural or stereoisomer, without further methods of separation.3 In either case, dehydration has poor selectivity and therefore it application is limited from mass producing single alkene isomers.
Alkene stereoselectivity is of key importance in a number of synthetic intermediates and target molecules, particularly ones which have medicinal applications, such as Panomifene, see Figure 1.4 The consequences of this need for stereoselectivity in synthetic routes has resulted in a number of olefination reactions being developed during the 20th century. The most prominent, useful and earliest stereoselective olefination being the Wittig reaction, which utilises a phosphorus-based ylide, as shown by Scheme 2.5
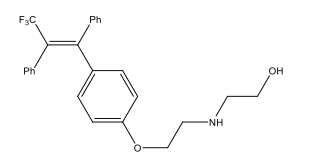
Figure 1: The antiestrogenic drug Panomifene; a superior drug to the well-known Tamoxifen. This is an example of a drug, whose pharmacological activity is dependent upon the stereochemistry of an alkene functional group.
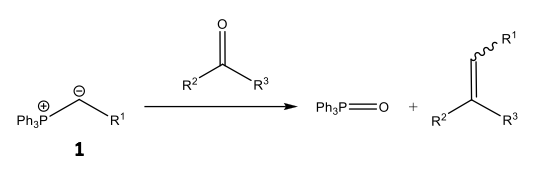
Scheme 2: An alkene synthesis via a generalised Wittig reaction has been presented. Note the use of a phosphorus ylide 1 and its ability to affect the stereoselectivity of the product alkene.
Wittig olefination (also known as the Wittig reaction) was developed by Georg Wittig in 1954; a reaction for which he was awarded a Nobel Prize in Chemistry 25 years later.6 Many other olefination reactions have been classified since Wittig’s publication, most of which involve a variety of heteroatoms substituting for the more commonly used phosphorus ylide, as well as some being mediated by transition metals.7
Peterson olefination, a commonly practised method of olefination, uses a silicon-based reagent to selectively convert an aldehyde or ketone into an alkene via a β-hydroxysilane precursor 2.8 A single diastereomer of the precursor may be stereoselectively converted into either cis- or trans-isomers via acidic or basic dehydration, respectively, as shown in Scheme 3.9
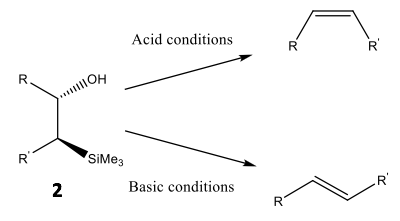
Scheme 3: The elimination reaction for an alkene precursor: a β-hydroxysilane diastereomer, under acid and basic conditions and the resultant stereoselectivity associated with said conditions.
Though Wittig olefination has been well characterised; many developments have been published since its discovery regarding in particular the mechanism, which is still open to discussion.10 At present, it is suggest that the carbanion end of the ylide attacks the aldehyde 3 and the resultant betaine then cyclises 4 to a stable oxaphosphetane 5. It is plainly obvious that the oxaphosphetane and its collapse must relate to the stereospecificity of the product 6, as shown in Scheme 4.11 It is not so obvious whether steric or electronic effects are more influential to the conformation of the betaine species, however, Vedejs correctly determined that the oxaphosphetane over betaine is the more crucial intermediate within the mechanism.12
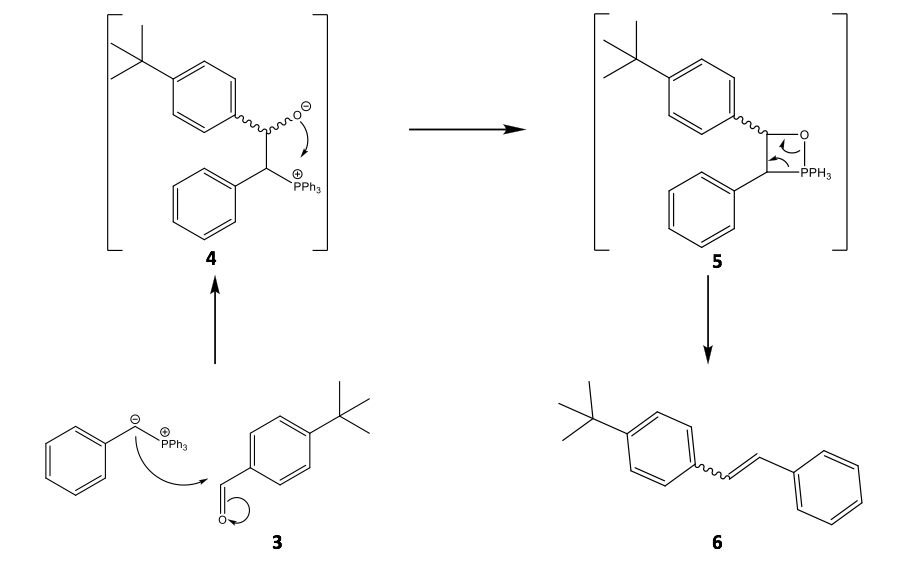
Scheme 4: The proposed mechanism of the Wittig olefination of 4-tertbutylbenzaldehyde with a partially stabilised phenyl ylide showing the possibility for either E- or Z-alkenes to form. This reaction is specific to the experimental method used within this report.
Wittig olefination stereoselectivity is complex, but can be generalised to the stability of the ylide. Stabilised phosphoryl anions will give E-alkenes, whereas non-stabilised anions will give primarily Z-alkenes. Even benzylic ylides, which can be considered partially-stabilised, will give E-alkenes.13 A possible explanation for this selectivity could relate to the oxaphosphetane or betaine formation being reversible, if the ylide is stable, and therefore only the faster of the two eliminations may occur to give the trans isomer.
Modifications, such as those described by Schlosser and Christmann, can be made for even more control on alkene geometry: forming the nonconventional E-alkene using a non-stabilised ylide, as shown by Scheme 5.14
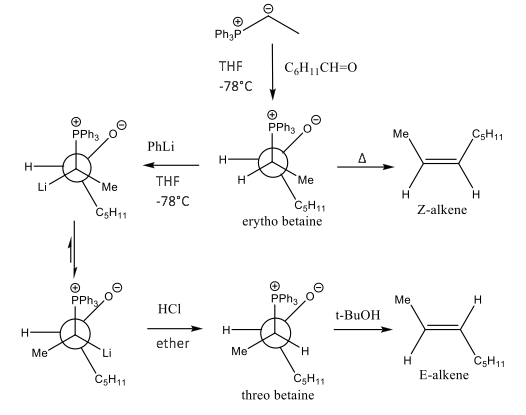
Scheme 5: This presents the Schlosser modification using the Klyne-Prelog conformer system. This modification enables the threo betaine species to form and therefore allow non-stabilised ylide to form E-alkenes. Change in group positions is dependent on the steric effects associated in the lithium adduct.
Method
Synthesis of ylide: A 3-necked round-bottomed flask was fit with stoppers and a condenser with a nitrogen inlet. The flask was filled with ethanol (75 ml) and then sodium metal (0.55 g, 24 mmol) was added over five minutes. The resulting solution was then heated to 30°C and benzyltriphenylphosphonium chloride (7.78 g, 20 mmol) was added over five minutes. The newly-made ylide solution was then cooled to 0°C until required for further use.
Synthesis of p-t-butylstilbene: A solution of ethanol (10 ml) and 4-tertbutylbenzaldehyde (2.92 g, 18 mmol) was then added to the ylide solution over one minute. The reaction mixture was then refluxed for 1.5 hours at ca. 105 °C. Work up was performed using hydrochloric acid solution (3 ml) and the solvent was evaporated in vacuo to afford a cream solid. The solid was triturated with petrol (15 ml) several times and all extracts were isolated via filtration and combined together. The petrol solvent was then evaporated in vacuo to afford a crystalline product. The product was then purified by recrystallisation using ethanol. This was followed by the purified solid being isolated via filtration and washed with ice-cold ethanol.
Results
p-t-butylstilbene: Yield 1.30 g (5.50 mmol, 31 %). m.p. 97.1 – 98.6 °C (lit.,15 trans isomer 96-98 °C) (lit.,16 cis isomer … °C). tlc Rf = 0.183 (1:1, ethyl acetate : hexane, on silica).
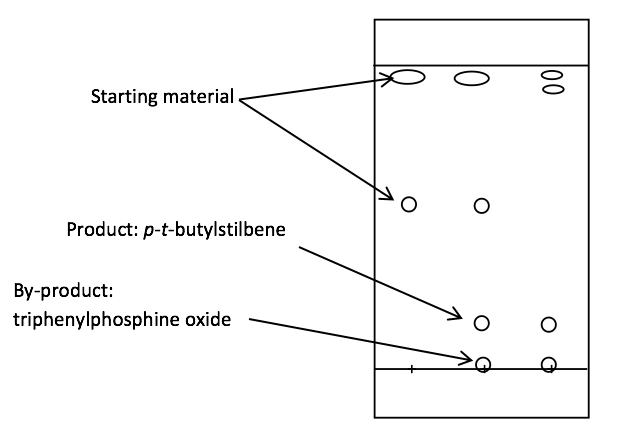
Figure 2: A thin layer chromatography plate showing three spots: purely 4-tertbutylbenzaldehyde, mixed and purely para-tert-butylstilbene from left to right.
I.R. vmax / cm-1: 3030 and 2950 sharp (=C-H and C-H aromatic), 1590 weak (C=C), 1570 weak (C-C aromatic).
1H NMR, (400 MHz; CDCl3; Me4Si), δ / ppm 1.4 (9H, comp., C(CH3)3), 7.12 (2H, d.d., 3J = 20.26 Hz, =C-H), 7.25 (1H, comp., C-H aromatic), 7.40 (4H, comp., C-H aromatic), 7.60 (4H, comp., C-H aromatic).
4-tertbutylbenzaldehyde:
I.R. vmax / cm-1: 3000 sharp (C-H aromatic), 2790 weak (H-C=O), 1720 very sharp (C=O, aldehyde), 1570 sharp (C-C aromatic).
n.b.: Please see appendix for raw data and spectra. Please also note, after frequent search, a literature value for the melting point of the cis- isomer was unable to be obtained.
Discussion
Hydrogen gas was evolved from the addition of sodium to ethanol. This effervesces was used as an indicator to whether the reaction had gone to completion on forming the in situ base: sodium ethoxide. A yield of 31% was … in comparison to other published articles, with similar methods.17
Thin layer chromatography showed that the reaction did not go to completion as indicated by a remainder of 4-tertbutylbenzaldehyde present in the third spotting, as shown by Figure 2. The chromatography also illustrated that product was being formed, since two new components can be seen: triphenylphosphine oxide and p–t-butylstilbene.
Infrared spectroscopy indicates a change in the functional groups present in the starting material and the products. Both fingerprint regions (<1500 cm-1) for the product and starting material are non-identical, indicating the formation of a new molecule. A single, sharp peak in the starting material spectrum at 1720 cm-1 indicates an aldehyde C=O stretch present in the original 4-tertbutylbenzaldehyde, which is not present in the spectrum for the product. Peaks in the product spectrum at 2950 and 1590 cm-1 represent =C-H and C=C stretches, which are not seen in the starting material (reference).18
All the points stated above indicate that a carbonyl, specifically an aldehyde functional group, has been converted into an alkene via the use of a phosphorus-based ylide. The conclusions that can be drawn from the infrared spectra concur with the chromatography data, that a new molecule has been formed, along with a very polar by-product being present in the TLC: most likely triphenylphosphine oxide.
Melting point analysis showed that the olefination reaction was successful, since the experimentally observed melting point corroborates with other published values. The melting point of 97.1-98.6 °C specifically matched the E-isomer literature melting point, in comparison to a much … Z-isomer melting point. It is typical for geometrical isomers to have variable physical properties (reference).19
Proton NMR analysis showed a doublet of doublets at 7.15 ppm, which was chemical shift characteristic of a deshielded, sp2 proton environment, i.e. an alkene proton (reference).20 The 3J coupling constant of 20.26 Hz is an indication of the vicinal protons being in an anti conformation with a torsion angle of 0° or 180° (the maximum), as described by the Karplus equation, see Figure 3 (reference).21 In other words, the vicinal coupling constant indicates that our product alkene is specifically the trans- diastereomer. The fact the NMR indicates that the product is the trans- diastereomer is also supported by the fact the melting point corresponds with the trans- diastereomer literature melting point and not the cis- diastereomer.
image
Figure 3: This illustrates both the Karplus equation and the effects of it being modelled with variable torsion angles (φ), which is equal to the vicinal coupling constant (reference).22
Conclusion
…Therefore, we may concur that our partially-stabilised benzyl ylide results in olefination to give trans- alkenes as the major diastereomer.
Briefly restate yield, m.p., tlc, ir, nmr results.
Do state whether you got E or Z isomer or a mixture, justified with the evidence you’ve collected.
Reflection
After reviewing the remarks on my previous report I have made considerable and minor changes to this report to improve both the style and content. Example of significant changes being: not using subscript for my reference numbering; trying to condense my writing in to a more concise format and focusing more on what the experiment was trying to achieve. I have also tried to not continually go out of third person during my report. In particular I will compare my conclusions to other published data to see whether this result has been repeated and therefore more reliable.
References
- S. Fordred, Ph. D. Thesis, University of Bath, 2012.
- S. Furniss, A. J. Hannaford, P. W. G. Smith, A. R. Tatchell, Vogel’s Textbook of Practical Organic Chemistry, Wiley, 5th edn., 1989, ch. 5, pp. 488
- Sojak, J. Hrivnak, P. Majer, Anal. Chem., 1973, 45, 293
- Konno, T. Daitoh, A. Noiri, J. Chae, T. Ishihara, H. Yamanaka, Org. Lett., 2004, 6, 933-36
- Wyatt, S. Warren, The Disconnection Approach, Wiley, 2nd edn., 2008, ch. 15, pp. 108
- Wittig, U. Schӧllkopf, Eur. J. Inorg. Chem.,1954, 87, 1318-30
- E. McMurry, Chem. Rev., 1989, 89, 1513
- J. Peterson, J. Org. Chem., 1968, 33, 780-84
- J. Ager, J. Org. React., 1990, 38, 1
- Vedejs, J. Org. Chem., 2004, 69, 5159
- Robiette, J. Richardson, V. K. Aggarwal, J. N. Harvey, J. Am. Chem. Soc., 2006, 128, 2394
- E. Maryanoff, A. B. Reitz, Chem. Rev., 1989, 87, 1318
- F. Silversmith, J. Chem. Ed., 1986, 63, 645
- Schlosser, K. F. Christmann, Angew. Chem. Int. Ed., 1966, 5, 126
- J. G. Smith, E. Oliver, T. J. Boettger, Organometallic., 1983, 2, 1577
Appendix
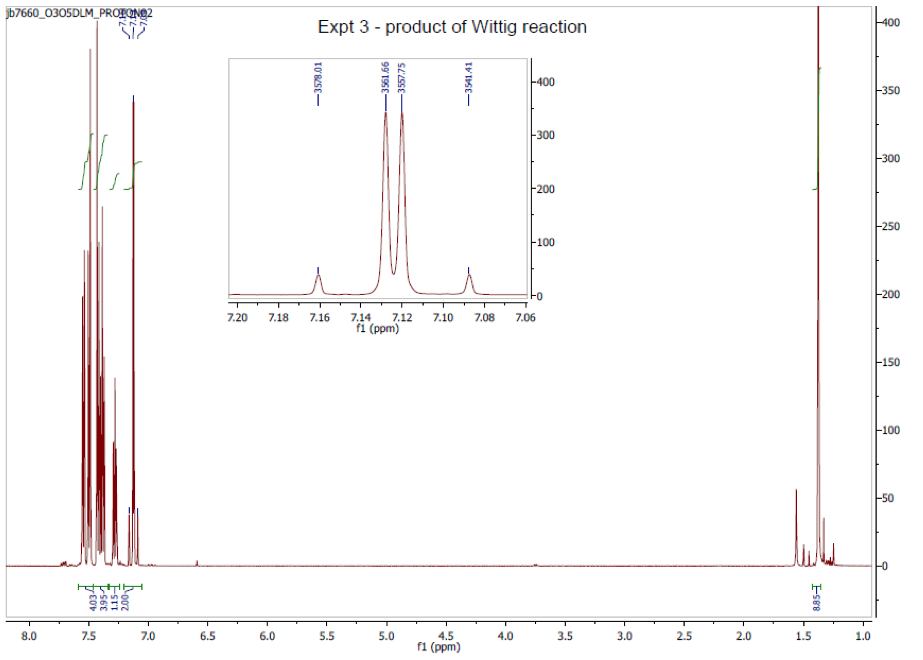
Figure 4: 1H NMR spectrum taken in deuterated chloroform and TMS at 400 MHz. Scan represents the product: p-t-butylstilbene.
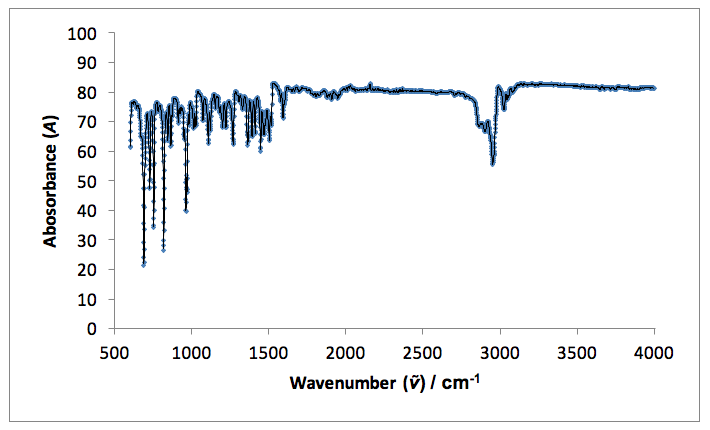
Figure 5: An infrared spectrum taken for the solid product: p-t-butylstilbene.
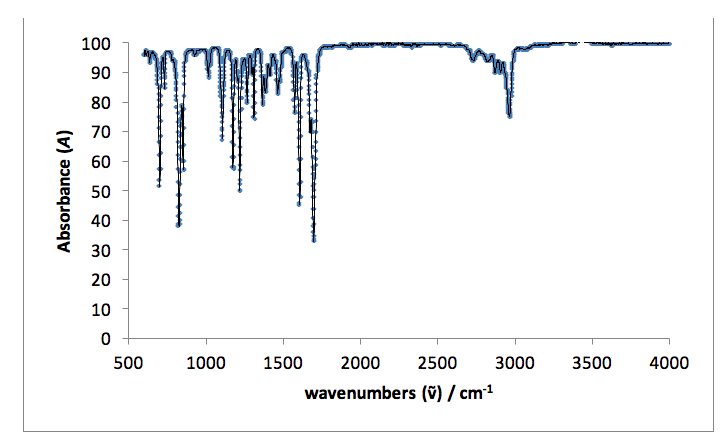
Figure 6: An infrared spectrum taken for the liquid starting material: 4-tertbutylbenzaldehyde.
