Preparation and investigation of organometallic compounds
Abstract
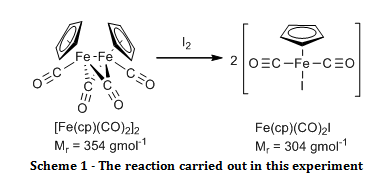
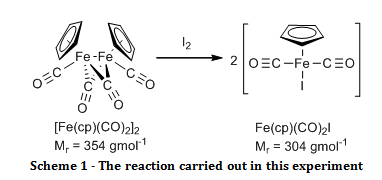
The organometallic compound, [Fe(C5H5)(CO)2]2, was oxidised to Fe(C5H5)(CO)2I and their infrared spectra were compared to show that there were bridging carbonyl ligands in the start material, but that they were absent in the product.
Introduction
Organometallic compounds are compounds that contain bonds between a metallic atom and a carbon atom. There are many applications of organometallic compounds which include use as catalysts (for example, the deuteration of organic compounds for use as NMR solvents is sometimes done by using (η5-cp)2TaH3)[i] or as reagents in synthetic routes (for example, a Grignard reagent, which have Mg-C bonds, are often used as nucleophiles to add alkyl groups to compounds). A common ligand in transition metal chemistry is carbon monoxide.
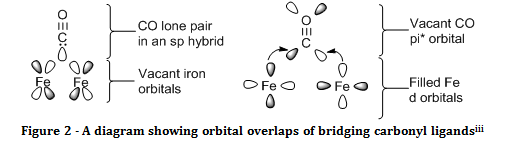
Metals that are bonded to carbon monoxide are often in low oxidation states, which mean that there can be an extra dimension to the way they form bonds with the carbonyl ligand. As well as the sigma bond formed by donation of carbon’s lone pair, there is back donation of electron density from the metal into the CO antibonding pi orbitals. This means that bonds between metals and carbonyl ligands tend to be strong and the compounds formed are fairly inert.
Evidence for back bonding is seen in the infrared spectra of compounds that contain carbonyl ligands. Donation into the CO π* orbital leads to the significant weakening of the CO triple bond. This means that the stretching frequency observed for metal carbonyl compounds is much lower than that of free carbon monoxide.
Carbon monoxide can also act as a bridging ligand between two metal centres. In the naming of a compound with a bridging ligand, the ligand is given the prefix of µ with a superscript number to show how many atoms the ligand bridges between, for example, µ3 CO would imply that there was a carbonyl ligand within the complex that is chemically bonded to three metal centres. The stretching frequency for bridging carbonyl ligands is usually shifted 100 – 200 cm-1 to lower wavenumbers than terminal carbonyls. This is due to the fact that back donation is felt from multiple metal centres, hence the CO bond is weakened to a much greater extent, and resonates at a much lower frequency.
The aim of this experiment was to oxidatively cleave the iron-iron bond in [Fe(C5H5)(CO)2]2 with iodine and compare the infrared spectra of the start material and the product, especially in the carbonyl ligand region.
Experimental
Solid [Fe(C5H5)(CO)2]2 (0.52 g) and iodine (0.63 g) were added to a two necked round bottom flask fitted with a nitrogen inlet, a reflux condenser and a nitrogen bubbler. Air was eliminated from the system by passing nitrogen gas through it for two minutes before adding chloroform (20 ml) and swirling until all of the solid had dissolved, at which point purging with the nitrogen continued. The reaction was brought to reflux for 30 minutes while maintaining a gentle blanket of nitrogen above the reaction throughout. Once cooled, the mixture was washed with aqueous sodium thiosulfate pentahydrate (4.0 g in 50 ml, 2 x 25 ml washes). The organic layers were combined and dried over sodium sulphate. The organic solvent was removed by rotary evaporation. The resulting black crystals were recrystallised from a 1:1 mixture of heptane and diethyl ether, washed with petroleum ether (60 – 80°C) (3 x 5 ml) and air dried to give the product Fe(C5H5)(CO)2I (0.895 g; 100 %; νmax/cm-1 3150 (C-H stretch, sp2), 1959 (C=O, terminal), 1423 (C=C, aromatic), 837 (Fe-I)).
Results and discussion
The theoretical yield for the reaction was 0.89 g, and since 0.89 g product were collected, the reaction gave a 100% yield. As much as it would be nice to have had a 100% yield, the product may still have been slightly wet, or there may have been traces of other reagents in the sample.
The IR spectra of the start material and the product were compared. The only significant difference between the two spectra was the absence of a peak at around 1750 wavenumbers in the IR spectrum of the product, which appeared as a sharp, prominent peak in the spectrum of the start material.
As stated in the introduction, the stretching frequency for bridging carbonyls is much lower than that of the terminal carbonyl by around 200 cm-1 due to the increased back donation onto bridging carbonyls. It can thus be perceived that, since there was a strong peak at around 1950 cm-1 in both spectra that were due to terminal carbonyl ligands, bridging carbonyl ligands were present in the start material, but not in the product. This makes perfect, logical sense, since the product formed only has one iron atom; hence there is nothing to bridge between.
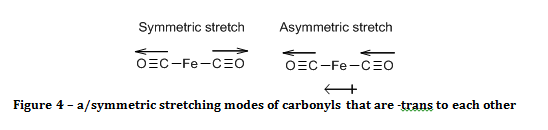
The spectrum of the product reveals that it must have a square planar, with the carbonyl ligands -trans to each other. This is known because there is only one CO band in the spectrum. The criterion for a bond stretch to be infrared active is that the stretch must result in a change in the electric dipole of the molecule. Since there are only two out of four carbonyl ligands in the product, there are only three possible structures. One possibility is that the product is tetrahedral, and the other options are that the product is square planar with carbonyl ligands either –cis or –trans to each other. The arguments against the structure of the product being tetrahedral or –cis square planar are the same.
With reference to group theory, the two carbonyls will act as a pair when they oscillate, and they will have a symmetric and an asymmetric stretch. When the carbonyls were –cis to each other (or in a tetrahedron) both of these stretching modes result in a change of the electric dipole of the molecule, as demonstrated in figure 3. As a consequence of this, two bands would be observed in the carbonyl region of the IR spectrum.
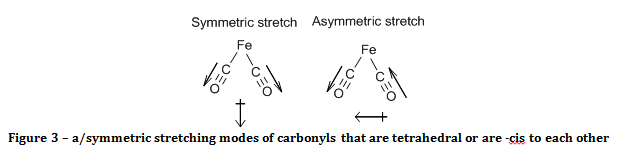
If the carbonyls were –trans to each other, then only the asymmetric stretch results in a change of the molecule’s electric dipole, as shown in figure 4, and hence is the only carbonyl peak observed.
The variable temperature 13C NMR spectrum for the start material, [Fe(C5H5)(CO)2]2, shows two peaks at low temperatures of -85 °C which slowly begin to coalesce at temperatures above -12 °C. This is due to the fact that the terminal and bridging carbonyl ligands exist in two distinct chemical environments, but are interchangeable, and the rate at which this happens is temperature dependant.
At temperatures greater than -12 °C, the rate of exchange is faster than the resolution of the spectrometer, and a single peak is observed with a chemical shift that reflects an average of the two chemical environments. Below -12 °C the rate of exchange is slower than the resolution of the spectrometer, so two peaks appear (one peak for each carbon environment).[i]
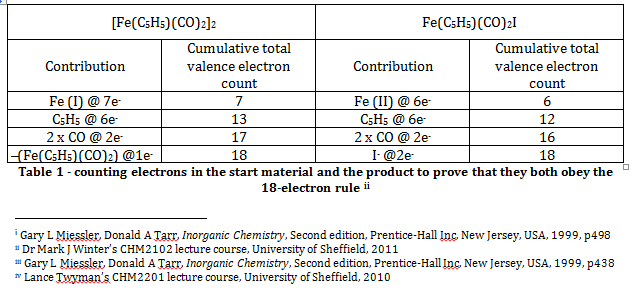
The Effective Atomic Number rule (or the eighteen electron rule) states that the total valence electron count for a transition metal complex should be equal to eighteen, and that the total electron configuration should be equal to that of the next noble gasii. Simple electron counting exercises for the starting material, [Fe(C5H5)(CO)2]2, and the product, Fe(C5H5)(CO)2I, show that both compounds obey this rule. Note that the C5H5 ligand is assumed to be a pentavalent ligand that utilises all six of its pi electrons for bonding with the metal centre.