Investigating Polyphosphates with 31P NMR Spectroscopy
Abstract
The experiment investigated the use of 31P NMR spectroscopy to determine the structure of polyphosphate anions. The structures of two cyclic (P4O124- and P3O93-) and two linear (P3O105- and P2O74-) were found.
Introduction
Phosphorus (V) has many polyphosphate oxo-anions which usually existing as either linear or cyclic phosphates. It is known that the linear phosphates are of the form [PnO3n+1](n+2)- and the cyclic phosphates in the form [PnO3n]n- (1). Two of these were synthesised: P3O105- and P4O124- using the following reactions:
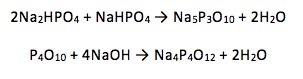
31P NMR is a well-known analytical technique which was used here to determine the structures of the polyphosphates under investigation. Polyphosphates have many uses, in nature and in industry. The very bases of life, RNA and DNA(2), rely on long phosphate chains linking nucleobases. This structure provides a strong backbone and allows the strands build up in only one direction. This is key for replication of cells. Biology also uses polyphosphates as a means of transferring energy. In the human body, adenosine triphosphate (ATP) is present in all living cells and is converted into adenosine diphosphate (ADP) which releases energy(2), without which the body could not live. They also have uses as sequestering agents which have such uses as water softeners as they are able to bind to metal cations in the water and so reduce such effects as limescale(2).
Results/Discussion
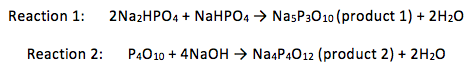
Table 1: A table of the properties of the reactants used and some of the products’ properties
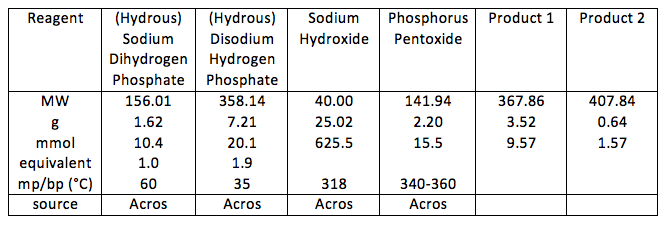
For the first reaction:
Before heating:
The mass of the crucible and lid without the mixture = 33.03g
with the mixture = 41.22g
=> the mass of the mixture = 8.19g
cf the combined mass of the reactants (shown above) = 8.83g
=> mass was lost during transfer to the crucible
After heating:
A white solid was formed (product 1).
The mass of the crucible, lid and product = 36.42g
Mass with product extracted = 32.90g
=> the mass of the product = 3.52g
The yield can then be calculated as follows:
Theoretical yield = number of theoretical moles(using the equation and the moles of the yield determining reactant) x GFM
= (20.1mmol/2) x 0.36786g/mmol
= 3.70g
%yield = (actual yield / theoretical yield) x 100
= (3.52g/3.70g) x 100
= 95.2%
The yield for product 1 is very high despite the loss of mass during the transfer to the crucible.
For the second reaction:
When the phosporus pentoxide was added to cold water a hissing sound was heard.
A white solid was formed when P4O10 was added.
The methanol started as colourless and clear but when the filtered reation mixture was added it became milky white and when sodium hydroxide was added it turned blue.
Mass of watchglass and product = 11.53g
Mass of the watchglass without the product = 10.89g
=> mass of product = 0.64g
Theoretical yield = 6.32g
%yield = 10.1%
Extremely low yield is most likely due to losing product by adding too much water to the methanol causing the product to dissolve in the water. Further loss could have been during the decanting off of the methanol/water mixture which may have still had some product in it. This loss could have been reduced by letting the product settle for longer before decanting.
NMR data
NMRs were run in deuterium enriched water at 121.5MHz, with a reference compound of CDCl3, 85% H3PO4
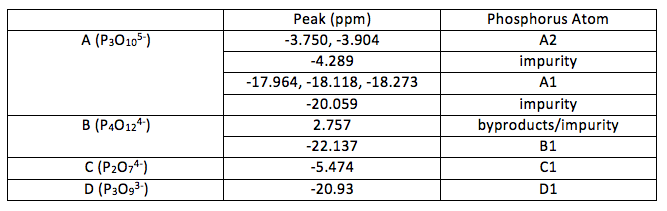
Table 2: Peaks in the NMR spectra and their associated atoms (as labelled in Figure 1)
From these results can be deduced the following structures:
Figure 1: Structures of the investigated polyphosphates, deduced using 31P NMR
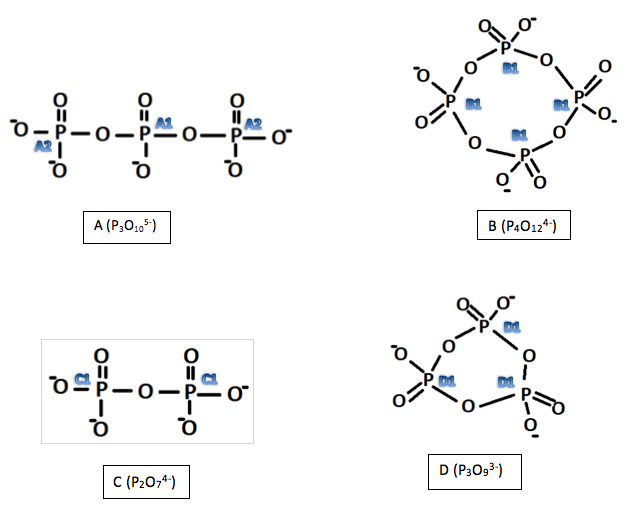
A1 was deduced as this would account for the environment that would have coupling effects from two nearby phosphorus atoms (1:2:1 triplet). A2 was deduced as each atom (equivalent to each other through symmetry) has only one phosphorus atom nearby to produce a 1:1 doublet. Looking at the relative volumes under the peaks this also fits with the elucidated structure although 31P NMR is not so reliable in quantative interpretations as proton NMR. The final peak is most likely due to any impurities in A which may be due to unreacted reactants or incomplete reactions. This peak is very small, suggesting that is not due to an atom in the molecule being investigated.
As there is only one main peak in spectra B it suggests symmetry makes all phosphorus atoms equivalent. This does not seem an unlikely deduction as the compound is expected to be cyclic. For a phosphorus (V) compound this makes the only likely structure the one shown in Figure 1. The small peak can be discounted as an impurity o some kind, possibly a by-product. The by-products formed in reaction 2 could be monophosphate anions made during the formation of B.
The occurrence of only one peak for C and D again suggests symmetry and equivalent atoms. Again, this is not surprising for D as it is cyclic. From this the structures of both could be determined.
From the structures deduced for A and C, the stoichiometry for hydrogen monophosphates used can be understood. A monophosphate would have the following structure:
Figure 2: Deduced structure of a monophosphate, using structures of the linear polyphosphates shown in Figure 1
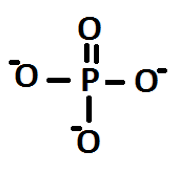
Here it is seen that there are three negative charges, each on an oxygen atom. In fact the charges would be delocalised over the four oxygen atoms:
Figure 3: The resonance structures of a monophosphate anion:
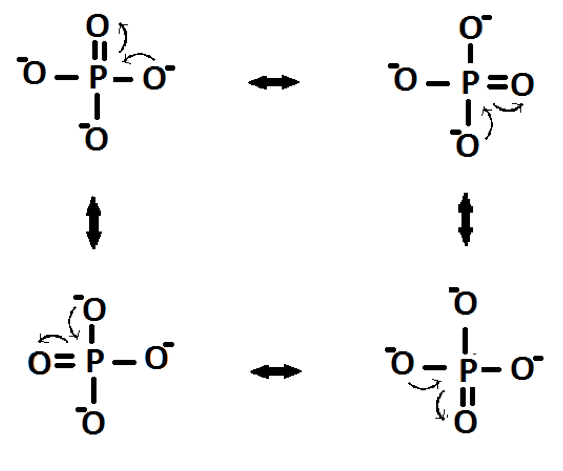
This structure accounts for the stoichiometry in that the anion needs three mono-charge cations to neutralise the charge. The mechanism for the formation of P3O105- is shown in Figure 4. It occurs through two separate nucleophilic attacks of oxygen atoms attacking the central phosphorus atom.
Figure 4: Reaction mechanism for the formation of P3O105-
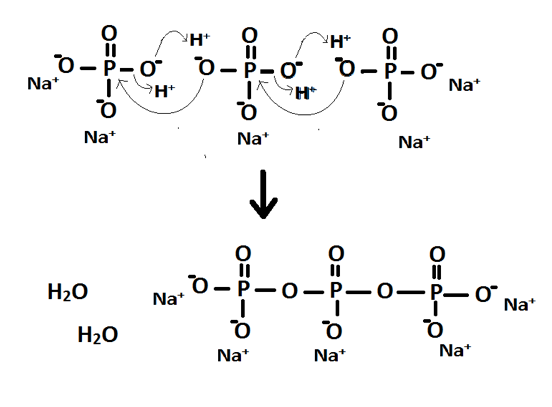
From this it can be seen that diphosphate could be formed instead in an incomplete reaction. This could account for the peak at -4.289 in spectra A however the spectra provided for diphosphate has a peak at -5.474 instead. If it had been over reaction and formed a polyphosphate with four or more phosphorus atoms then it would occur as a series of peaks that would show coupling involved. The peak may simply be impurities of another kind.
The structure deduced for P4O124- is similar to that deduced for the structure of P3O93- which is understandable as they are both members of the same family of cyclic polyphosphates. Phosphorus pentoxide was used as a synthetic precursor as it provided a framework from which, through hydrolysis, a four member phosphate ring could be formed. By-products could include the phosphate anion or P3O93- but these are not seen in the NMR spectra. The very small peak that cannot be associated with the P4O124- molecule does not relate to either of these however and so other by-products must also be possible.
Experimental(1)
Equipment and general Techniques
The main analytical technique used was 31P NMR. This used a Bruker av300.
Safety
Care was taken to ensure that phosphorus pentoxide was not inhaled as it is very corrosive and produces a fine dust which is easily inhaled. For this reason all procedure involving the chemical was carried out in a fume cupboard. Sodium dihydrogen phosphate, bromothymol blue and sodium hydroxide are all corrosive and care should be taken when handling them. Methanol is very toxic and very flammable and so should be kept away from any naked flames or sources of ignition and should not be allowed to come into contact with skin.
Procedure
Hydrous sodium dihydrogen phosphate (1.62g) and hydrous disodium hydrogen phosphate (7.21g) were heated together at 540-580°C for two hours. The white powder (product 1) was then run through 31P NMR. The mass of the product was found to be 3.52g.
Phosphorus pentoxide (2.20g) was gradually added to cold water such that the temperature remained between 5-10°C. Once the white solid formed had dissolved, the solution was neutralised using a sodium hydroxide solution (25% w/v) and bromothymol blue as an indicator. The solution was filtered and added to briskly stirred methanol (300cm3). This turned the methanol from colourless to milky. Sodium hydroxide solution was again added until the methanol solution had just turned blue. Water was added until the precipitate started clumping together. Once the product (product 2) had settled, the liquid was decanted off before the product was vacuum desiccated over fresh phosphorus pentoxide. A free flowing powder was not produced and so the powder was ground in methanol. The methanol was then removed using a rotavap, giving a 0.64g, free flowing white powder. This was then tested using 31P NMR.
In both cases the products were made suitable for NMR using deuterium enriched water.
References
(1). University of St Andrews CH2501 laboratory manual, 2013-2014
(references Vogel’s textbook of practical chemistry, Vogel et al, Longman, 1989 (5th edition); Practical organic chemistry, J.T. Sharpe et al, Chapman and Hall, 1989; Experimental organic chemistry, Harwood et al, Blackwell 1989 (1st edition) 1998 (2nd edition); Practical skills in chemistry, Jones et al, Prentice Hall, 2002)
(2). C.E. Housecroft and A.G. Sharpe, “Inorganic Chemistry”, 3rd edition, 2008
