Effects of Substituents on the Absorption Spectra of molecules
Written by John
Introduction:
The objective of this lab was the observation of the effects of substituents on the absorption spectra of molecules (most specifically azulene). ChemDraw 3D was used to visualize the electron densities and energies of the HOMO and LUMO states of several molecules. Then, the absorption spectra of several chemicals—including azulene, mono-substituted azulenes, and poly-substituted azulenes—were obtained using spectrophotometers, and these absorption spectra were compared. These spectra allowed the explanation of shifts in HOMO and LUMO states in context with Electron Donating Groups and Electron Withdrawing Groups.
Molecular Orbital theory plays an important part in this experiment. This theory, which involves the Linear Combination of Atomic Orbitals, is the idea that the electron orbitals of a molecule are different from those of the separate atoms. This suggests that the molecules have their own electron orbitals, which can be found by combining the atomic orbitals mathematically with the Schrödinger equation. These molecular orbitals fill much the same way as atomic orbitals do: only two electrons in each orbital, opposite spin. 1
The molecular orbitals on the outside of the molecule are called the Frontier Orbitals. These orbitals, the HOMO and LUMO, often play a large role in reactivity and absorption/transmission spectra. The HOMO is the Highest Occupied Molecular Orbital, which means the highest orbital in which electrons are found in the ground state. The LUMO is the Lowest Unoccupied Molecular Orbital, which is the electron orbital just above the HOMO. Because of the electronic interaction between these two orbitals, one of the most effective ways to evaluate these orbitals is the use of spectrophotometry. The maximum wavelength absorbed during these tests marks the energy difference between these two orbitals. 2
The molecules observed in this experiment were aromatic molecules. Molecules exhibit aromaticity when they possess alternating double bonds in a cyclic pattern. These double bonds tend to represent electrons which are delocalized, meaning that they traverse the whole of the molecule. Aromaticity describes how the structure in question is stronger and more stable than the conjugation of the bonds would inherently suggest. 2
Two molecules of this experiment actually have the same molecular formula (C10H8), but different structures. These isomers, naphthalene and azulene, exemplify a significant difference in structure. Naphthalene is known as a benzenoid structure. These molecules are those resembling the structure of benzene, which features six-carbon rings. Azulene, on the other hand, consists of one ring of five and another ring of seven carbon atoms. This makes azulene a non-benzenoid molecule. 2
The two substituents which were included in this experiment are known as Electron Donating Groups and Electron Withdrawing Groups. These behave in much the way their names suggest. Electron Donating Groups (or EDGs) donate electron density to the molecule to which they are attached. This leads to a less stable state, and a higher energy level for the orbital involved. Electron Withdrawing Groups (EWGs) withdraw electron density from the molecule to which they are attached. This leads to a more stable state, and a lower energy level for the orbital involved. 1
An important concept to know when considering substituents with a molecule such as azulene is how the placement of these substituents affect the molecules. The mnemonic device HOLE is used to this effect: HOMO Odd, LUMO Even. This means that a substituent which is attached to an odd carbon affects its HOMO; a substituent attached to an even carbon affects its LUMO. This paired with the explanations of the effects of EDGs and EWGs can help with the understanding of the shift in absorption spectra seen in the experiment.
Procedure:
Week 1:
ChemDraw 3D was used to model naphthalene, benzene, azulene, and anthracene. Their energies were minimized using MM2 within the program, and their HOMO and LUMO energies were recorded. Pictures of each molecule were saved and sketched. Additionally, the differences of the HOMO and LUMO energies were determined.
ChemDraw 3D was used again to model the azulene molecules with substituents: 1-nitroazulene, 4-isocyanoazulene, 5-formamidoazulene, and 2-aminoazulene. Pictures were both sketched and saved to the computer for future reference. Their energies were minimized and HOMO and LUMO energies recorded. The differences of these energies were again found.
A spectrophotometer was connected to the computer and LoggerPro software opened. The spectrophotometer was calibrated using a “blank” cuvette containing ethanol. Then, the absorption spectra was collected and saved for the following molecules: naphthalene, anthracene, and azulene. These molecules were also drawn.
Four cuvettes were then obtained, containing 1-nitroazulene, 4-isocyanoazulene, 5-formamidoazulene, and 2-aminoazulene. Their Lewis structures were drawn, then their colors recorded and their absorbance spectra obtained using the spectrophotometer. The lowest energy peak, or max wavelength, of the absorbance spectra was recorded, and pictures of each spectrum were saved for future reference, and then sketched. The effects of the substituents on the azulene molecules was discussed. (See Conclusion)
Week 2:
A spectrophotometer was connected to a computer and LoggerPro software opened. The spectrophotometer was calibrated using a “blank” containing ethanol, and then was used to find the absorbance spectra of each of the following chemicals:
6-amino-1,2,3-triphenyl azulene
6-formamido-1,2,3-triphenyl azulene
6-isocyano-1,2,3-triphenyl azulene
6-methyl-1-nitroazulene
4,6,8-trimethyl-1-nitroazulene
1,3-azulenyldicarboxylic acid
5-isocyano-1,3-di-tert-butylazulene
The absorbance spectra were saved for future reference, as well as sketched. Prior to the use of the spectrophotometer, predictions were made concerning the color shift the solution would exhibit (red or blue). Also, the max wavelength and the actual shift was recorded for each of the compounds.
The effects of the functional groups on their respective azulene’s HOMO and LUMO orbitals were discussed, and comparisons were made of several of the functional groups.
Table 1 The maximum wavelengths of the absorptions of several chemicals.
|
Compound |
λmax |
|
Naphthalene |
310 nm |
|
Anthracene |
375 nm |
|
Azulene |
690 nm |
Table 2 The wavelengths and subsequent wavelength shifts of mono-substituted azulenes in comparison to a non-substituted azulene.
|
Compound (Structure) |
λmax |
λmax shift (relative to azulene) |
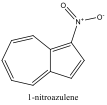 |
590 nm |
Blue Shift |
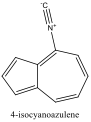 |
730 nm |
Red Shift |
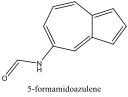 |
722 nm |
Red Shift |
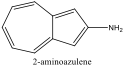 |
484 nm |
Blue Shift |
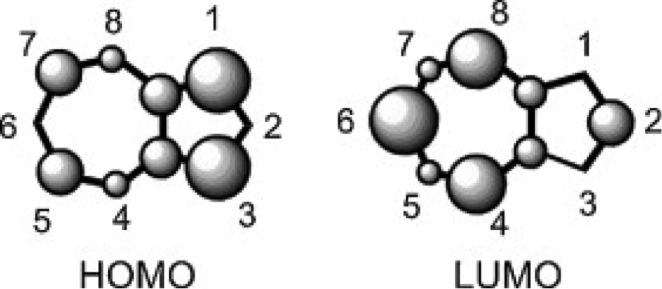 Figure 1 The relative electron densities of the HOMO and LUMO orbitals of a non-substituted azulene molecule.3
Figure 1 The relative electron densities of the HOMO and LUMO orbitals of a non-substituted azulene molecule.3
Table 3 The results of Week 2 of the experiment, involving poly-substituted azulenes
|
Compound (structure) |
λmax |
Predicted λmax shift (relative to azulene) |
Observed λmax shift (relative to azulene) |
 6-amino-1,2,3-triphenyl azulen480 nm 6-amino-1,2,3-triphenyl azulen480 nm |
480 nm |
Red |
Blue |
 6-formamido-1,2,3-triphenyl azulene 6-formamido-1,2,3-triphenyl azulene |
675 nm |
Red |
Blue |
 6-isocyano-1,2,3-triphenyl azulene 6-isocyano-1,2,3-triphenyl azulene |
>800 nm |
Red |
Red |
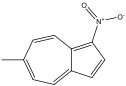 6-methyl-1-nitroazulene 6-methyl-1-nitroazulene |
570 nm |
Blue |
Blue |
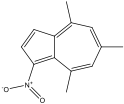 4,6,8-trimethyl-1-nitroazulene 4,6,8-trimethyl-1-nitroazulene |
490 nm |
Blue |
Blue |
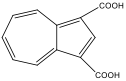 azulene-1,3-dicarboxylic acid azulene-1,3-dicarboxylic acid |
580 nm |
Blue |
Blue |
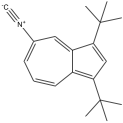 5-isocyano-1,3-di-tert-butylazulene 5-isocyano-1,3-di-tert-butylazulene |
790 nm |
Red |
Red |
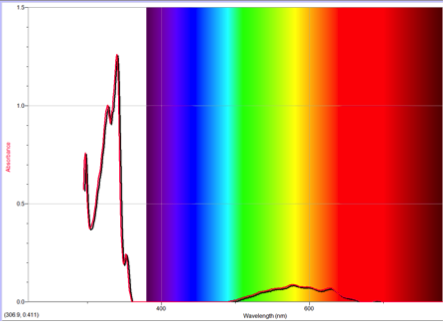
Figure 2 Absorption spectrum of azulene
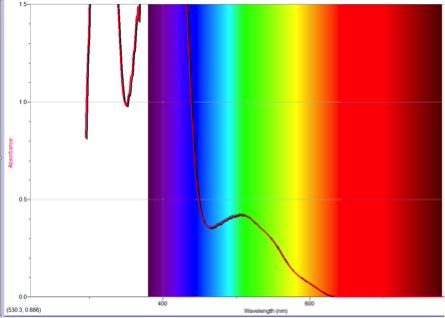
Figure 3 Absorption spectrum of 1-nitroazulene
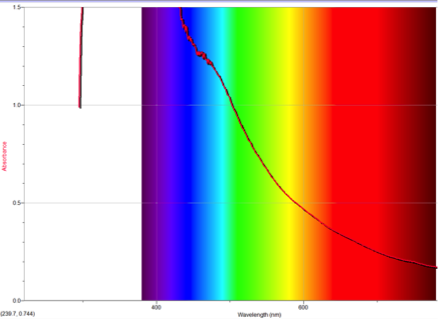
Figure 4 Absorption spectrum of 4-isocyanoazulene
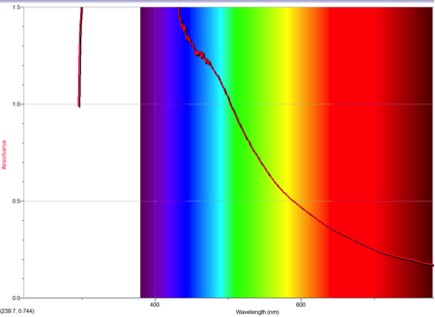
Figure 5 Absorption spectrum of 5-formamidoazulene
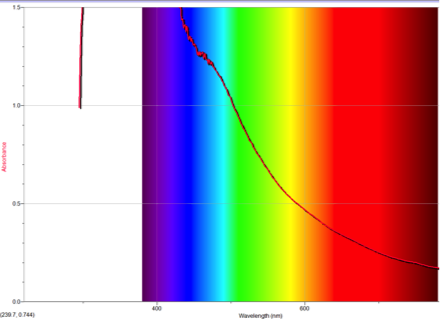
Figure 6 Absorption spectrum of 2-aminoazulene
Discussion:
One of the many important differences between naphthalene and azulene is their HOMO and LUMO electron densities. Naphthalene’s electron densities lie along similar planes in different molecular orbitals. This means that the electrons of naphthalene experience more repulsion from each other than those of azulene, whose HOMO and LUMO densities alternate between odd and even placements (Figure 1). This increased repulsion causes an S0-S1 gap whose emitted wavelength falls outside human vision.
Substituents alter the emitted wavelengths of these molecules by affecting the electron densities. Electron Withdrawing Groups (or EWGs) withdraw electron density, thereby making the arrangement more stable. The energy level of the orbital affected subsequently lowers, causing a typically-noticeable red shift in the maximum wavelength. Alternately, Electron Donating Groups (EDGs, for short), add to the electron densities of the molecule. This causes the affected orbital to be less stable, and requires more energy to obtain. The orbital affected depends on the placement of the substituent. A useful mnemonic device in this matter is HOLE: HOMO Odd, LUMO Even. This explains that the affected orbital alternates between odd and even placements.
The absorption of these molecules with substituents is consequently affected. A substituent which would cause the S0-S1 energy gap to close (such as an EWG on even or an EDG on odd) would therefore cause a red shift. A substituent which would cause the S0-S1 energy gap to widen (such as an EWG on odd or an EDG on even) would consequently cause a blue shift. Therefore, it may be assumed that the expected changes in absorption may be explained by the following table:
Table 4 A summarization of the effects of certain substituents on the spectra based upon their placement.
|
EWG |
EDG |
|
|
ODD |
Blue shift |
Red shift |
|
EVEN |
Red shift |
Blue shift |
To understand this concept, an evaluation of the absorption spectra would be helpful. Consider Figures 2, 3, and 4 (Results section), which compare the absorption spectra of a non-substituted azulene and an azulene molecule with Electron Withdrawing Groups on either an odd or even position. Note that the spectrum of 1-nitroazulene possesses a blue shift, while the spectrum of 4-isocyanoazulene experiences a red shift in comparison to the spectrum of non-substituted azulene. (See Table 1 for maximum wavelengths of “original” molecules.)
Alternately, Figures 2, 5, and 6 (Results section) display the effects of Electron Donating Groups on different positions. The spectrum of 5-formamidoazulene experienced a red shift while the spectrum of 2-aminoazulene experienced a blue shift.
These results, which can be found in table 2 coincide with the expected ones based upon the understanding of HOMO/LUMO relationships with EDGs and EWGs. The HOMOs of molecules with substituents on odds lower for EWGs and raise for EDGs. Similarly, the LUMOs of molecules with substituents on evens also lower for EWGs and raise for EDGs.
These effects can be compounded within a single molecule, as in the case of seven of the multi-substituted azulenes in the experiment. Table 3 shows related data. Explanations for the effects of each of these molecules follow.
6-amino-1,2,3-triphenyl azulene experienced a blue shift, which contradicted the prediction of a red shift. However, this result is explainable. The phenyl substituents, which were EDGs, on odds would have forced the HOMO up, while the phenyl substituent on even would have pushed the LUMO up as well. The amino is also an EDG, which would have pushed the LUMO up, additionally. The blue shift suggests that the energy gap increased, so the LUMO was affected more strongly. This means that the formamido must be a stronger EDG than phenyl.
6-formamido-1,2,3-triphenyl azulene also experienced a blue shift, which negated the red shift of the prediction. This, however, is perfectly in line with what the concepts this lab cover. The two EDG phenyl’s on odd would force the HOMO up, while two EDGs (one phenyl and one formamido) on even would also force the LUMO up. Since there was a blue shift, it can be inferred that the energy gap increased. This means that the LUMO was affected more, suggesting that formamido is a stronger EDG than phenyl.
6-isocyano-1,2,3-triphenyl azulene experienced a red shift, which confirms the corresponding prediction. The two EDG phenyls would cause the HOMO to move up; one EDG phenyl on even would force the LUMO down while the one EWG isocyano oneven would force the LUMO up. Since one phenyl on odd and one phenyl on even “cancel out”, the remaining forces cause the levels to move closer. This corresponds with a red shift.
The phenyl group of multisubstituted azulenes offers some interesting comparisons. Because the maximum wavelength of 6-amino-1,2,3-triphenyl azulene is 480 nm and the max wavelength of 6-formamido-1,2,3-triphenyl azulene is 675 nm, it can be assumed that the effect of the amino is stronger than that of the formamido. The energy difference was greater, causing a more significant blue shift. Because isocyano is a withdrawing group, it had a smaller gap, causing a red shift, and is therefore more difficult to compare qualitatively with the other two.
6-methyl-1-nitroazulene experienced a blue shift, which corresponds with the prediction. The EDG on even would force HOMO down while an EWG on odd would force LUMO up, leading to a larger energy gap. This corresponds with the blue shift in the spectrum.
4,6,8-trimethyl-1-nitro azulene experienced a blue shift, which corresponds with the predicted shift. An EWG on odd would cause the HOMO to shift down while three EDG on even would cause the LUMO to shift up. This leads to a much larger gap between HOMO and LUMO, which would account for the blue shift that was observed.
4,6,8-trimethyl-1-nitroazulene has more EDGs, so the blue shift was noticeably higher. The shift observed led to a wavelength max of 490 nm, while 6-methyl-1-nitroazulene’s max wavelength was only 570 nm. The larger blue shift of 4,6,8- suggests that the effects of the EDGs compounded.
1,3-azulenyldicarboxylic acid experienced a blue shift, which is consistent with the prediction. Because two EWGs on odd positions would cause the HOMO to lower, the energy gap between the HOMO and the LUMO would increase. This translates into the observed blue shift.
5-isocyano-1,3-di-tert-butylazulene experienced a red shift, which corresponds with the red shift of the prediction. The two EDGs on odd positions would cause the HOMO to raise while the EWG on odd would cause the HOMO to lower. Because a red shift was observed, this suggests that the HOMO increased (and the energy gap between the HOMO and LUMO closed). Given this, the tert-butyls are stronger than the isocyano.
This experiment provided some opportunity for error. For one, a miscalibration of the spectrophotometer or the misreading of the spectra could have led to false data, which would alter results by providing incorrect shifts. Also, it isn’t absolutely certain that the cuvettes used had only pure substances inside. It’s possible that they were contaminated with other chemicals, which would alter the absorption spectra, as well. This would also adversely affect the shifts observed.
Conclusion:
The goal of this experiment was the observation and explanation of the effects of substituents on azulene. For this purpose, the HOMO and LUMO of several comparable molecules were observed using ChemDraw 3D. Then, the absorption spectra of several mono- and poly-substituted azulenes were collected, and the shift in maximum wavelengths used to achieve this end.
Spectroscopy was an acceptable method of testing the effects of substituents on azulene for a number of reasons. First, it would enable the observation of the maximum wavelength absorbed, which indicates the energy difference of the HOMO and LUMO of the molecule in question. Comparisons using these maximum wavelengths are relative, but relatively easy to make, which also led to its use.
One of the most important parts of this experiment is the understanding of substituents’ effects on the HOMO and LUMO of azulene. Electron Withdrawing Groups stabilize these orbitals, which results in a lower energy. Electron Donating Groups destabilize these orbitals, which results in a higher energy. If a substituent is placed on an odd carbon, then the HOMO is affected; if a substituent is placed on an even carbon, then the LUMO is affected.
These points are used in the explanation of the observations. For example, if a chemical’s absorption experiences a red shift after a substituent was added, there are two possibilities: an Electron Withdrawing Group was placed on an odd carbon, or an Electron Donating Group was placed on an even carbon (in a mono-substituted azulene). For a blue shift, the opposites must be true: an Electron Withdrawing Group was placed on an even carbon, or an Electron Donating Group was placed on an odd. Therefore, if the chemical’s name—which reveals the substituents position—was known beforehand, as in this experiment, the absorption spectra allows a quick assessment of whether the substituent was an Electron Withdrawing Group or an Electron Donating Group.
The case becomes slightly more complicated in the second part of the experiment involving poly-substituted molecules. However, this portion of the experiment provided the substituents’ identities (EWG or EDG), so what remained was the determination of which interactions were present. In the case of the phenyl solutions, a comparison was made of the remaining substituent. The same goes for the methyl/trimethyl solution.
It was found in part one of the experiment that nitro and isocyano were Electron Withdrawing Groups, and formamido and amino were Electron Donating Groups. These were found by comparing the shift in absorption spectra to that of “plain” azulene. A blue shift on odd and a red shift on even (as in the first instance) suggested an Electron Withdrawing Group. A red shift on odd and a blue shift on even (as in the second) suggested an Electron Donating Group. These results and understandings contributed to the second portion of the experiment.
Several of the predictions of the color shift of the poly-substituted azulenes were correct; however, the first few were incorrect. This was an error in understanding of the experimenters, who were factoring the S2 energy level (or LUMO+1) in the prediction, which should have been discounted. Otherwise, the understanding of the concepts and the knowledge of which groups were EDGs and EWGs in part two of the experiment to largely successful predictions. Several important observations include the following: amino is a stronger EDG than phenyl, formamido is a stronger EDG than phenyl, tert-butyls are stronger groups than an isocyano, and overall, the effects of EDGs and EWGs are compounded within a molecule.
Works Cited
- “Tuning Color Through Substitution”. Handout.
- Oxtoby, David W., Gillis, and Campion. Principles of Modern Chemistry. 7th ed. Philadelphia: Saunders College Pub., 1986. Print.
- Mikhail V. Barybin, Nonbenzenoid aromatic isocyanides: New coordination building blocks for organometallic and surface chemistry, Coordination Chemistry Reviews, Volume 254, Issues 11–12, June 2010, Pages 1240-1252, ISSN 0010-8545, http://dx.doi.org/10.1016/j.ccr.2009.11.002.
